

Harren, Tobias等人于2022年在Journal of Chemical information and Modeling上发表了一篇“Interpretation of Structure–Activity Relationships in Real-World Drug Design Data Sets Using Explainable Artificial Intelligence”。下面我们来做一下翻译和解读。
基于深度神经网络 (DNN) 的计算机模型有望用于预测新分子的活性和特性。不幸的是,它们固有的黑箱特性阻碍了我们对哪些结构特征对活性有重要影响的理解。然而,这些信息对于捕获潜在的结构-活性关系 (SAR) 以指导进一步优化至关重要。为了解决这一解释上的差距,“可解释人工智能”(XAI)方法最近开始流行。**在此,我们将多种XAI方法应用于具有完善的SARs和可用的X射线晶体结构的线索优化数据集项目,并进行了比较。**我们可以看到,通过将DNN模型与一些强大的解释方法相结合,可以获得易于理解和全面的解释。**特别是,基于 SHAP 的方法有望完成这项任务。**使用基于原子的热图的新的可视化方案提供了对基础SAR的有用见解。值得注意的是,所有的解释只有在基础模型和相关数据的背景下才有意义。
1.引言
寻找新型的、强效的、耐受性好的分子作为特定靶蛋白的活性物质,最终应产生新的候选药物用于临床开发。在设计符合特定目标特征的新分子时,必须以多参数的方式考虑许多不同的方面。在此,采用现代计算机方法的计算机辅助药物设计(CADD)可以极大地指导项目团队完成这一任务。
如今,先进的机器学习技术被常规地应用于从实验数据中开发统计模型,以指导进一步的化合物设计。这些模型的不同应用领域包括通过生成性人工智能(AI)方法进行的从头设计、合成预测和逆合成分析以及分子特性的预测。这些特性预测包括ADMET(吸收、分布、代谢、消除、毒性)和理化特性(如logP或溶解度),以及靶蛋白的生物活性(酶和细胞活性)或抗靶点。
随着人工智能和深度学习方法的日益普及,深度神经网络 (DNN) 经常被用作属性预测的高级统计引擎。术语 DNN 是指由具有一个以上隐藏层的神经网络组成的用于预测的复杂非线性统计模型。更快的计算机硬件、改进的算法以避免神经网络过度拟合,加上许多计算机平台上的软件解决方案和改进的训练算法,促进了DNN在人工智能领域的成功应用,例如计算机视觉、语言处理以及药物设计,如无数成功的例子证明了这一点。
虽然一些研究探索了 DNN 方法相对于经典机器学习 (ML) 方法的潜在优势,但 DNN 的一个缺失特征是对结果模型的解释或“可解释性”。大多数基于人工智能的机器学习方法本质上都是作为黑匣子构建的,算法的设计通常不允许理解为什么某种方法会产生某种预测。因此,人们无法探索为什么会得到好的或错误的预测,也无法有针对性地改进方法。如果不能提供结构方面的解释,用户(如药物化学家)的接受程度往往较低,而可解释的预测可能会通过激发对先导结构的新化学转化为前瞻性设计提供更好的指导。
该方法涉及通过系统地去除或添加小的取代基对结构进行增量修改。根据 Polishchuk 等人的工作,对分子和虚拟匹配对的结果预测用于估计小替代对预测特性的影响。其中还提供了对模型解释的经典方法的回顾。
目前对这一问题的解决方案采用了与模型无关的方法来检测重要的结构特征,如基于结构碎片和衍生化的 "反应图 "来检测和可视化局部属性梯度。这种方法包括通过系统地去除或增加小的取代基来逐步修改结构。根据Polishchuk等人的工作,对分子和虚拟匹配对的预测结果用于估计小取代物对预测性质的影响,其中也有关于模型解释的经典方法的回顾。
现在,有几种方法被引入,以从深度神经网络中获得模型解释。这些方法包括,例如,像LIME或SHAP这样的框架,它可以为具有固定规模输入的神经网络提供解释,以及更普遍的方法,例如,基于梯度(gradients)或反向传播(backpropagation)。基于梯度的方法通过计算一个特征的变化对最终预测的影响来提供解释。相比之下,基于反向传播的方法是将输出传播回输入值,以估计这些特征的影响。最近,描述了用于解释基于图形的神经网络的综合梯度可解释人工智能(XAI)方法,并用于解释四个ADMET相关数据集。到目前为止,药物设计中的应用主要集中在提取分子中对某一特性有贡献的单一特征,以及最近将分子的某些部分标记为阳性或阴性,而没有关于贡献的相对强度的信息。然而,相对强度和贡献对于仔细调整分子的活性或分析分子修饰的影响至关重要。我们试图通过使用基于原子的热图对结果进行新的可视化来填补这一空白,该热图允许将获得的解释定量映射到分子上。
这些解释DNN预测的方法还没有被应用于未来的药物设计项目中,以使结构-活性关系(SAR)合理化。任何有用的解释都应首先正确预测两种相关化合物的活性趋势(例如,匹配的分子对,即一个小取代基与另一个小取代基的交换),然后根据观察到的活性变化清楚地认识到这一不同特征的重要性。此外,有关重要结构特征的信息应该是容易获得和理解的,以便为改善生物活性或其他特性的新的化学想法得出结论。
虽然有经验的药物和计算化学家经常手动对小数据集或匹配的分子对执行这些分析,以便为下一个优化周期提供依据,但生物和结构数据的庞大数量往往使这种分析无法用于更大和结构更多样化的数据集。
因此,我们研究的目的是探索不同的方法来解释针对两种具有药理学相关性的靶蛋白,即丝氨酸蛋白酶Xa(fXa)和天冬氨酸蛋白酶肾素的生物活性(蛋白质-配体结合亲和力Ki和IC50)而训练的深度神经网络的预测。由于这两个靶标的SAR是有据可查的,而且这两个靶标的蛋白质-配体复合物的几个X射线结构已经得到解决,这为探索基于DNN的统计模型解释是否能够捕捉SAR信息提供了一个很好的例子。
为了应用这些方法,我们首先在Xa因子和肾素的中型SAR数据集上训练了预测性DNN模型,然后实施不同的方法来解释神经网络预测。最后,我们提出了一种基于热图的方法,可以将模型解释定量地映射到分子结构的二维描述上,并支持在分析所有实验数据的基础上为下一轮的合成设计新的类似物。特别是与早期的工作相比,热图的使用大大促进了方法的应用,因为它不仅提供了关于单一的、抽象的特征的信息,还提供了结合所有特征的提取信息的单一解释。
2.材料和方法
2.1 神经网络模型
我们探索了两种类型的DNN模型,它们被证明对药物化学数据集最感兴趣,即使用二维指纹作为输入向量的简单多层感知器和图卷积神经网络(GCNNs)。后一种网络方法是基于Duvenaud等人的工作,**而我们应用了Ramsundar等人的DeepChem实现。**为了生成基于指纹的网络模型,我们使用了RDKit中的Morgan圆形指纹,它与著名的扩展连接性指纹(ECFP)高度相似。
摩根指纹使用中心原子周围一定大小的环状邻域来表示化学二维结构。他们的特征代表了一个特定的子结构的存在,并且没有预先定义,所以他们可以包括大量的结构特征。对于典型的机器学习应用,分子中的所有子结构被折叠成一个固定大小的向量,形成机器学习的输入指纹。
GCNN也在化学子结构上工作;但它们不是使用固定的输入特征列表,而是通过反复应用图卷积操作来提取相关的子结构,从而积累关于特定原子邻域的信息。这些信息在最后的密集层中被压缩成一个固定大小的向量,但是神经网络训练的本质允许为给定的任务选择相关的子结构信息,因此预计会产生一套更适合机器学习问题的描述符。我们最近展示了GCNN模型在药物设计应用中的良好表现,这促使我们采用这样一种方法。
2.1 解释神经网络的方法
解释神经网络的预测并不是一件简单的事情。神经网络本身是不透明的,必须应用特定的算法来提取其中的信息。在本节中,我们将介绍一些方法,我们已经探索了将这些方法用于理解我们的输入活动数据集。由于某种特定的方法并不自动适用于任何神经网络架构,因此并非所有的方法都能最终应用于所有的网络类型。
原子热图
原子热图是一种概念上简单的方法,将输入分子的单个原子转化为虚拟原子,并监测预测活性的变化。更具体地说,我们使用原子编号为 0 的 RDKit 原子来完成这项任务。**因此,单个原子与模型预测的相关性是通过将原始预测与修改后分子的预测进行比较来估计的。**此前,Sheridan对此进行了探讨,并显示出对所使用的描述符和机器学习技术的强烈依赖。我们对这种方法的研究只是作为本工作中讨论的数据集的比较器。
分层相关性传播(LRP)
这种方法基于获取网络输出并通过网络传播回来的想法。在每个神经元上,计算每个输入对神经元输出的影响。通过一直传播到输入层,可以计算出每个特征对输出的贡献程度。在相关研究中,类似的方法已经应用于 GCNN。在参考文献24中,使用了激励反向传播,它更适合于分类任务。在参考文献23中,应用了LRP,尽管是在一个不同类型的GCNN上,它是基于一个稍微不同的实现。
由于以指纹向量为输入的网络(如ECFPs)比较简单,其方法可以从原始工作中直接拿来进行应用。下面的方程式是针对GCNN的。为了解释这个方法,我们考虑由 N 个原子组成的单个输入分子 X。根据现有的方程,我们必须推导出 GCNN 使用的层类型的规则。首先,我们考虑卷积层。这些可以被认为是包含3个隐含层:局部总和池化、密集层和总和(Local Sum Pooling、Dense Layer和Total Sum)。局部总和池化层计算与中心原子相邻的所有原子的atomwise特征之和。在密集层中,权重被应用于累积的邻域特征,以及中心原子的特征。在总和部分,这些中心原子和邻域原子的特征被加上,并应用一个激活函数。在下面的公式中,R指的是一个相关性,l是一个层的索引,而ni和nj是一个层内神经元的索引,
z
n
i
n
j
z_{ninj}
zninj是沿着这些神经元之间的连接传递的值(输入乘以权重),而znj是所有这些连接的总和。因此,
R
n
i
←
n
j
(
l
,
l
+
1
)
R_{ni←nj}^{(l,l+1)}
Rni←nj(l,l+1)是第l层的神经元i与第l+1层的神经元j的连接的相关性,而
R
n
j
l
+
1
R_{nj}^{l+1}
Rnjl+1是所有来自神经元j的连接的相关性之和。

Dropout和Batch normalization层只包含输入到输出的1:1映射,因此它们保留了相关性。

剩下的两种类型是池化层。Graph-Pool应用局部最大池化。因此,它们的整个输出仅取决于一个输入,并且整个相关性被传递回单个原子的单个神经元。Graph-Gather层应用全局sum pooling和全局max pooling。对于max pooling,与之前的情况相同。对于sum pooling,我们有一个未加权的总和。
梯度热力图
解释神经网络的一个典型方法是梯度热力图法。在相关的报告中,这些方法已经被应用于GCNN。在这里,我们使用他们的表达方法作为baseline来定义我们的梯度热力图。这种方法的基础是输出y相对于输入x的梯度。

如果网络的输入是一个指纹,其结果是一个向量,其中包含分子子结构的相关性。**在GCNN的情况下,计算出的梯度是一个向量,包含了单个原子特征与输出的关联性。**正负部分将被相加,以得到一个原子的总正负贡献。
SHAP
另一组用于模型解释的方法是 “沙普利加法运算法”(SHAP)。这些方法是基于博弈论中的Shapley值。它们都是可加特征归因方法,并共享几个有用的属性(局部准确性、缺失性、一致性)。Bajorath等人对分子描述符做了更详细的介绍,我们只做一个介绍,进一步的细节请参考以前的工作和公开的实现。我们使用了SHAP的三种方法,即Kernel-SHAP、Gradient-SHAP和Deep-SHAP。**这三种方法都要求输入是固定大小的张量。因此,它们只能应用于以指纹向量为输入的模型。**受LIME启发的Kernel-SHAP,通过对输入进行小的改变并分析这些改变的效果来提供解释。根据这些,来估计单个输入特征的相关性。
梯度-SHAP基于输入,使用输出的梯度来估计特征的相关性。Deep-SHAP基于DeepLIFT,使用反向传播算法,追踪输出是如何根据输入产生的。梯度-SHAP和深度-SHAP都只在神经网络上工作。
用于可视化模型解释的热图
我们的目标是从DNN模型中得出相关的模型解释,这些解释与已知的结构-活性关系信息一致,并且容易理解,对进一步的先导化合物优化有明显的影响。以前的研究大多通过检查提取的最重要特征来验证他们的解释。虽然这提供了初步的SAR信息,但我们的目标是直接和全面的解释。为此,我们使用热图代表主要的视觉方法,用于展示和交流DNN模型的解释。其中一些方法已经计算出模型的特定原子相关性或重要性,可以直接映射到这些原子上。其他方法为化学子结构生成相关性。对于这些情况,我们使用化学子结构进行基于SMARTS的子结构搜索,并将特征相关性添加到子结构的每个匹配原子上。这就导致了通过计算所有匹配值的总和,形成了一个原子特异性的表征。我们选择添加所有的相关性,而不是应用例如最大运算,因为几个可能有负面和正面相关性的特征,可能包含一个单独的原子,只有这些特征的组合才能提供一个整体的原子相关性,在这个过程中,互补的影响被平衡,类似的影响被累积。类似地,为了仅仅增加相关性,可以通过独特子结构的大小来扩展它。图S1中显示了一个例子。可以看出,对于基于SMARTS的分布来说,得到了一个更平滑的着色,可以进行更精细的区分。然而,由于两者都很容易实现,根据使用情况和数据集的不同,其他着色方案也可能是有用的。
图S1 将SHAP的相关性添加到原子的不同方案。上图:指纹的SMARTS匹配,并将相关性添加到所有原子中(默认)。下图:通过将每个相关性除以片段的大小来分配相关性。
为了有效地比较图像与热图和二维化学结构,我们总是将提交分析的整个系列的分子的色标归一(图1)。此外,通常在色标上应用70%的截断值(最大颜色强度为最大数值的70%),这样可以更好地进行视觉区分,特别是在低值区域。
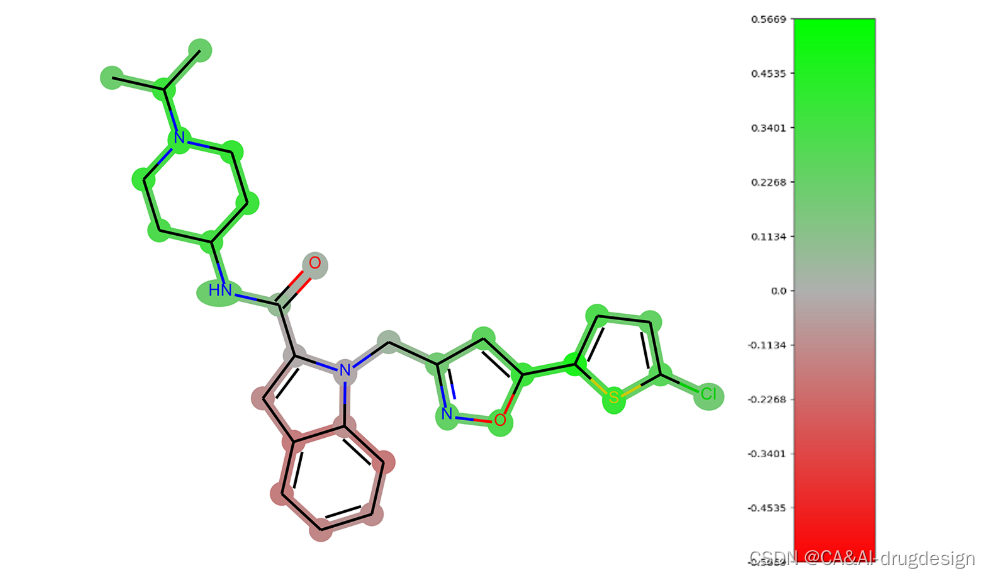
图1 从PDB文件2boh中提取的fXa抑制剂的热图可视化实例。一个带有截断值的热图与相应的颜色条显示。绿色数值代表对活性有利的结构特征,红色数值代表不利的特征。
对于计算单个子结构的相关性的方法,我们还对相应的虚拟模型预测产生的最积极和最消极影响的特征进行视觉化。虽然这些信息较少,但它们也很容易理解。
数据集
所有虚拟模型的建立、预测和解释都是利用三个数据集进行的,这些数据集包含了针对两个具有药理学意义的蛋白质靶标,即肾素和fXa的化学结构和活性。由于对这两个靶标都进行了内部项目,因此可以获得来自单个实验室的、直接反映蛋白质-配体相互作用的一致的生物数据。此外,对于每个系列,都解析了多个X射线结构,其中一些可在蛋白质数据库(PDB)中找到。这些数据集是药物设计项目中产生的数据集的实际例子。它们被故意地没有被进一步加强,以遵循人们在真实世界的药物设计项目中所面临的挑战。总共产生了三个数据集(下文有更详细的描述)。**两个数据集(肾素和围绕fXa的一个系列的子集)代表了小的数据集,包含一些高度相似的化合物和一些探索的变体。第三个数据集(所有fXa化合物)包括几个不同的系列,因此更加多样化。**图2给出了fXa数据集中所覆盖的化学空间的可视化图。由于决定在一轮设计中应合成哪些分子总是由前一轮的结果所指导,一些变体可能会更频繁地出现,因为它们导致了性能的改善,而一些变体则大大降低了活性,没有被再次使用,从而导致了数据集中的单一离群数据点。
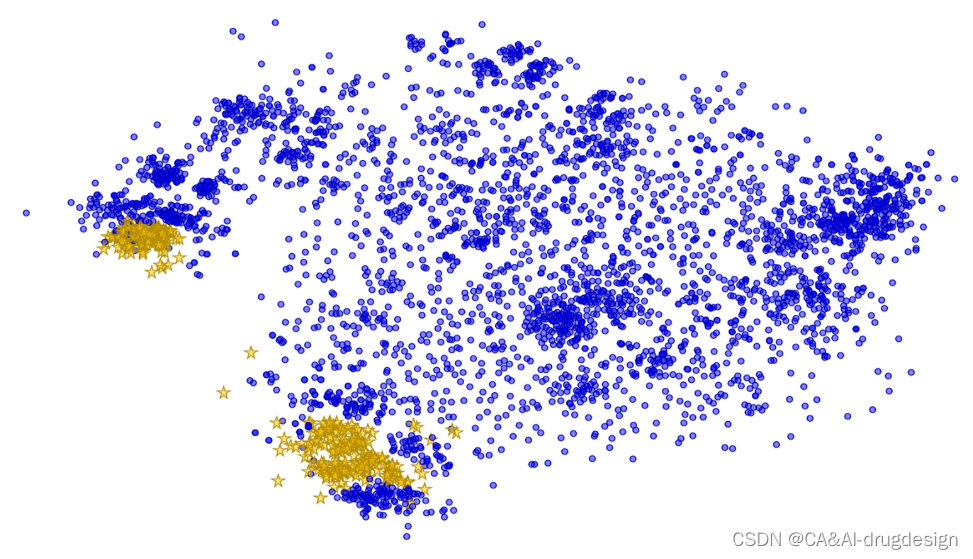
图2. 整个fXa数据集的相似性图。吲哚系列的子集用橙色的星星突出显示。相似性图是用D360中实现的UNITY指纹计算的,相似性阈值为70%。
对于天冬氨酸蛋白酶肾素,共探讨了142个吲哚-3-甲酰胺和氮杂吲哚的数据集。所有的化合物在文献中都有报道,生物活性以pIC50值给出。一个吲哚-3-甲酰胺(来自参考文献41的化合物5k(图3),IC50=2 nM)的代表性X射线晶体结构的PDB文件为3oot(2.55 Å分辨率)。其他X射线晶体结构在进一步的出版物中披露。在X射线结构分析和基于结构的设计的指导下,吲哚和氮杂吲哚骨架周围的各种位置被装饰上了一系列的取代物。这个小但典型的工业先导化合物优化数据集的SAR被很好的建立了。
对于丝氨酸蛋白酶fXa,我们研究了两个不同大小和结构多样性的数据集。生物活性以pKi值提供。第一个数据集由吲哚-2-甲酰胺系列的398个fXa抑制剂组成,分为训练集、验证集和测试集,分别为318个、40个和40个分子。**这个系列是以前在文献中报道过的,其特点是在吲哚N1位置针对蛋白酶S1口袋和在吲哚C2位置针对S4口袋的适当装饰。**此外,在中心吲哚骨架上还探索了几个取代物。这又包括一个典型的先导优化数据集。PDB文件2boh(2.20 Å分辨率,图4)显示了这个系列的一个有效成员(Ki=3 nM)的代表性X射线晶体结构。fXa的第二个数据集是一个大型的内部数据集,包括总共3950个分子,分为训练、验证和测试集,大小为3160、395和395个分子,代表我们内部药物发现项目的多个化学系列。这个数据集的DNN模型以前在文献中报道过,并被用作基于人工智能的从头设计的奖励函数。**采用这种较大的异质性数据集的一个重要方面是研究基础数据的规模和多样性对最终模型解释的影响。**特别令人感兴趣的是,来自化学多样性或重点数据集的模型之间的解释性会如何不同。生物学数据和参考X射线结构与较小的数据集相同。
模型训练
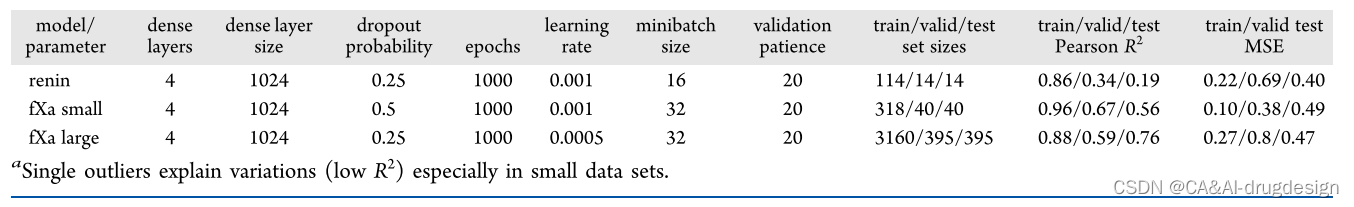
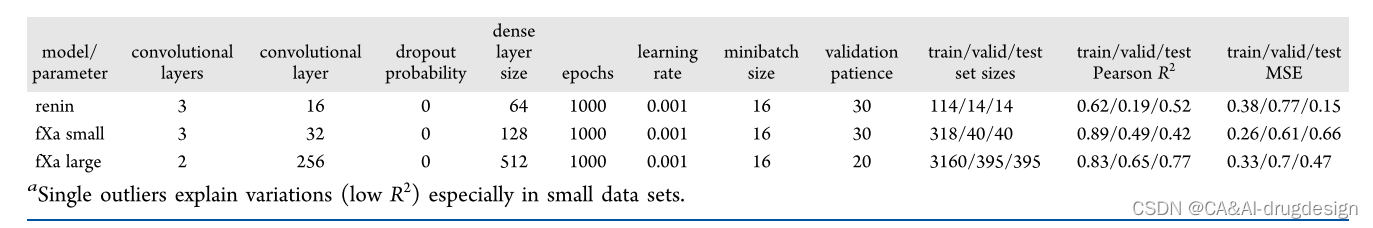
为了探索使用不同的XAI方法进行模型解释的潜力,我们首先为所有数据集训练了DNN模型,如上图所示,使用指纹或GCNN featurization。对于每个模型,使用RDKit中的MinMax算法将数据集划分为训练、验证和测试集。然后对神经网络的不同超参数进行参数优化。我们首先最大化了验证R2值。由于这本身不一定能提供最佳模型,我们还考虑了MSE(平均平方误差)来选择最终的最佳模型。对于指纹模型,需要优化的参数有密集层的数量和大小、dropout概率、epochs数量、学习率、最小批次大小以及validation patience。对于GCNN,这些参数是图形卷积层的数量、神经指纹的密集层的大小、epochs数、学习率、最小批次大小、dropout概率以及validation patience。如果loss或验证R2在validation patience给定的步数内没有变化,则用早期停止的方法停止训练。然后,每个方法和数据集的最佳模型被用于所有进一步的分析(见表1和表2)。
表1. 基于指纹的模型的参数和测试性能a
结果和讨论
为了成功地应用于命中化合物和先导化合物的优化,任何可解释的机器学习模型都应正确反映由较小的结构变化引起的亲和力改变趋势。随后的模型解释最好能捕捉到这些与亲和力有关的重要趋势,并使之可视化。任何有用的解释都应该与某一特定系列的已知SAR相一致。这种SAR信息可以从R-group分析、匹配分子对分析或蛋白质-配体复合体的代表性X射线晶体结构中提取。为了解决这个问题,我们在虚拟计算模型中分析了fXa和肾素数据集中的几个例子,其中小的结构变化导致了大的亲和力差异。
肾素蛋白-配体的相互作用
在图3中,显示了作为肾素抑制剂数据集成员的一个典型的吲哚甲酰胺的结合模式和基本的蛋白质-配体相互作用(PDB:3oot,2.5 Å分辨率,化合物5k,IC50=2nM)。这个X射线晶体结构显示了一个 "closed-flap闭合瓣 "的蛋白质构象,正如在许多其他的肾素-配体复合物中观察到的那样(例如阿利吉仑,PDB:2v0z)。哌嗪的二级胺参与了与天冬氨酸Asp32和Asp215的离子氢键相互作用,而邻近的羧酰胺氧则与Thr77-OγH相互作用。两个配体的苯基环都有利地参与了与肾素S1和S3子口袋的亲脂性相互作用。2-甲基-3-氟苄基基团用氟最佳地填补了一个小的、亲脂性的子口袋中的S1口袋。氟和Asp32的羧酸盐之间的附近也算作一个正交的多极作用。2-甲基基团朝向由Gly217Cα、Val30Cγ和Phe117环组成的亲脂性区域。此外,在S1苯环和Tyr75之间观察到一个有利的edge-to-face类型的CH-π-π作用。吲哚的取代基也有利地影响了结合亲和力,特别是5-OH基团,它与His287的咪唑形成氢键,而4-甲基取代基与Thr77Cγ接触。
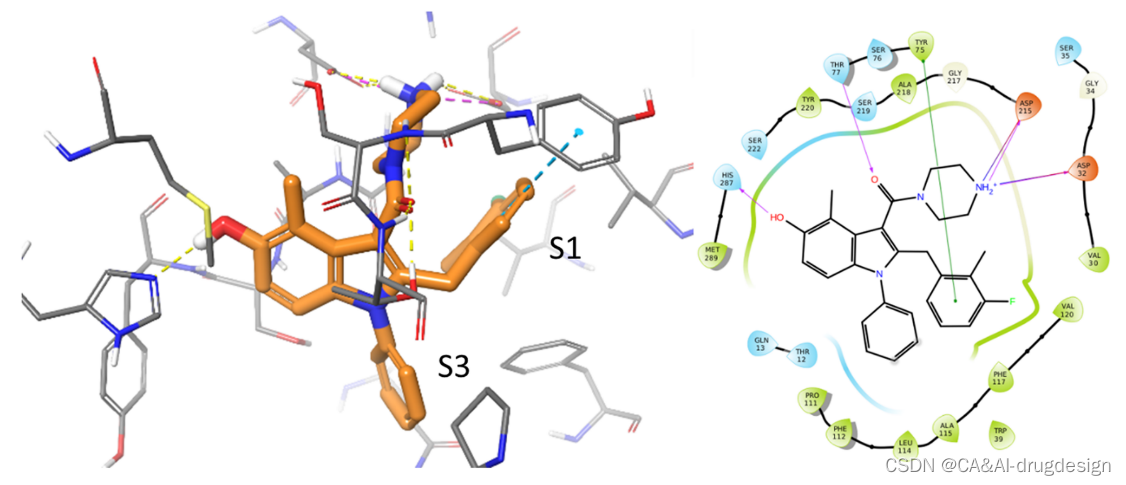
图3. 基于结构优化产生的具有代表性的吲哚-3-甲酰胺(橙色碳原子,2.5 Å分辨率,PDB:3oot, IC50=2 nM)与人肾素复合物的X射线晶体结构。灰色的碳原子显示了蛋白质的结合部位。关键的蛋白质-配体相互作用表示如下:黄色的氢键,青色的π-π相互作用,紫色的盐桥。所有的三维图都是用Maestro程序生成的。
最重要的相互作用之一是由哌嗪环中带正电的氮形成的。然而,由于这个结构元素在这个系列的优化程序开始之前就已经知道了,在数据集中没有这个motif的重大变化。因此,我们不会期望任何监督模型能突出这一区域的重要性,尽管这一点从X射线结构中是已知的。在吲哚的N1和C2位置,探索了许多有可能产生疏水相互作用的取代基。正如检查X射线结构所预期的那样,不同大小的取代基会显著影响结合亲和力。在吲哚的C5至C7位置,结构上允许小的取代物,其中一些取代物,如图3所示,形成额外的相互作用并提高活性,而其他取代物则有相反的效果,特别是如果它们太大。
Factor Xa蛋白-配体的相互作用
对于fXa,代表性的吲哚-2-甲酰胺1的X射线结构(分辨率:2.2 Å,PDB:2boh,Ki=3nM)可用于了解该特定系列的SAR。在图4中,显示了该系列成员的实验性结合姿势和蛋白质-配体的相互作用。与其他许多fXa的X射线结构一样,1的氯噻吩基团被埋在蛋白酶S1口袋中,氯指向Tyr228的芳香环。这个卤素很好地取代了其他含苯甲酰胺的fXa结构中的一个保守的水,并参与了与Tyr228的π-系统的氯-π相互作用。
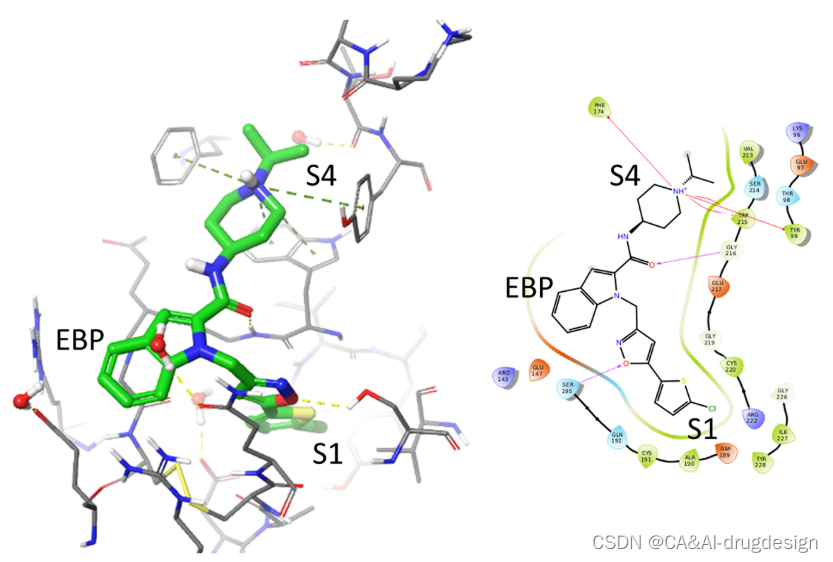
图4. 一个有代表性的吲哚-2-甲酰胺(绿色碳原子,2.2 Å分辨率,PDB:2boh,Ki=3 nM)与人类Factor Xa复合物的X射线晶体结构。灰色的碳原子显示了蛋白质的结合部位。水分子用红色的氧球表示。详见图3。
这种结合模式与其他在S1中含有氯的配体相似,并已被报道用于各种不同的配体。噻吩硫接近Val213-Cγ和Trp215-Cα,而其碳原子则朝向S1底部的Asp189侧链。异噁唑与活性位点Ser195之间有氢键相互作用,这种氢键作用是由Ser195中的Oγ和异噁唑的氮原子和氧原子形成。在这个复合物中没有观察到其他氢键。吲哚-2-羧酰胺的羰基氧离Gly216-N太远,不可能有强的相互作用,而可以推测是结构上保守的水桥连接了两个伙伴。
异丙基哌啶取代基结合在S4口袋中,被来自Phe174、Tyr99和Trp215等氨基酸的芳香环包围,形成这个口袋。疏水相互作用在这个口袋中占主导地位,**但由于潜在的有利的阳离子-π相互作用,碱性氮原子也被容纳在Trp215环系统的顶部。**在这个口袋边缘的氢键受体网络或结构上保守的水不可能有进一步的极性作用。
吲哚苯环位于Cys191-Cys220二硫桥的顶部,形成 “酯结合袋”(EBP),该口袋由Gln192、Arg143和glu147的柔性侧链衬托。Gln192侧链和吲哚的电子密度表明具有灵活性flexibility。
机器学习模型
这里描述的所有数据集的ML模型都显示出中等性能,主要是由于训练数据集的规模相对较小。尽管如此,它们在大多数情况下确实捕捉到了重要的SAR趋势,这对于设计进一步的实验往往是有用的。验证或测试集的肾素模型观察到的R2值较低,这是由于整个系列分子的规模(大小)有限,这些子集中的个别分子含有未在训练集中出现的子结构。然而,这些motif有时与显著的活性差异有关,被称为 “活性悬崖”。通常情况下,不会再合成含有已知对活性有不利影响的子结构的化合物,特别是对于基于结构的设计项目。关于模型性能的更多详细信息,请参考支持信息,在那里我们提供了模型的散点图(图S2-S4)。这些表明,只有单一的离群点导致了看似很低的R2值,因此,这些并不能正确反映实际的模型性能。
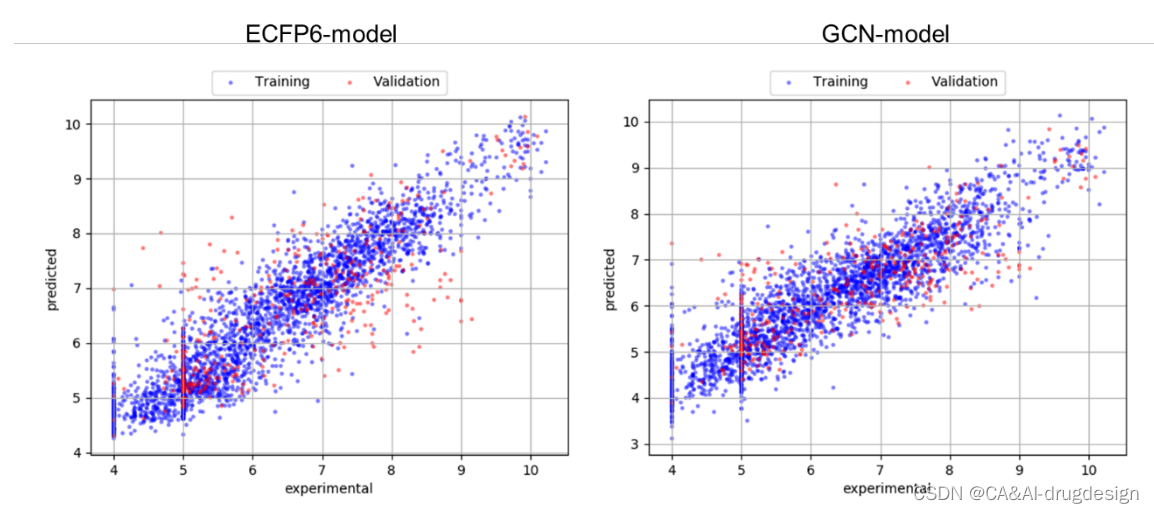
图S2 ECFP4模型(左)和GCN模型(右)的训练和验证集的预测与实验数据的散点图,来自大型fXa数据集。
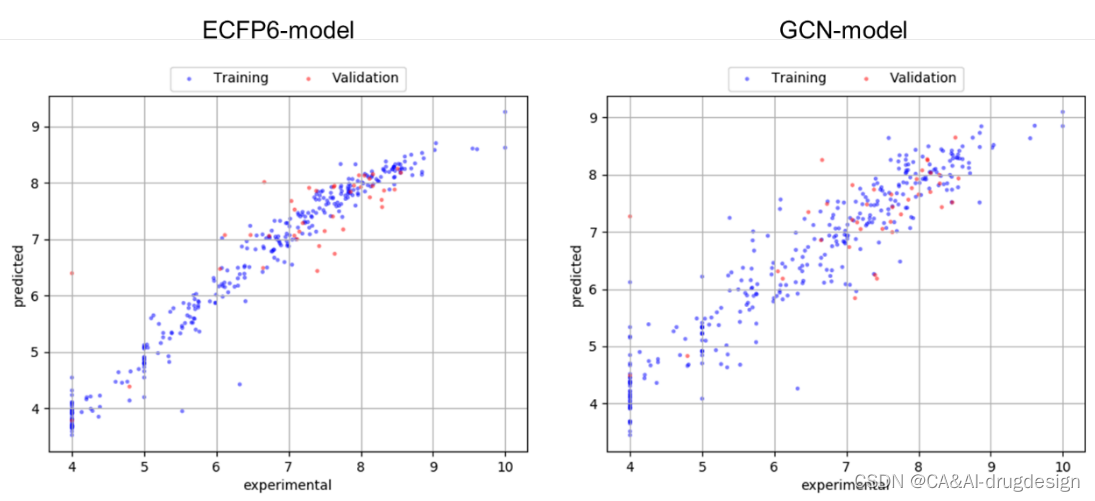
图S3 ECFP4模型(左)和GCN模型(右)的训练和验证集的预测与实验数据的散点图,来自小型fXa数据集。
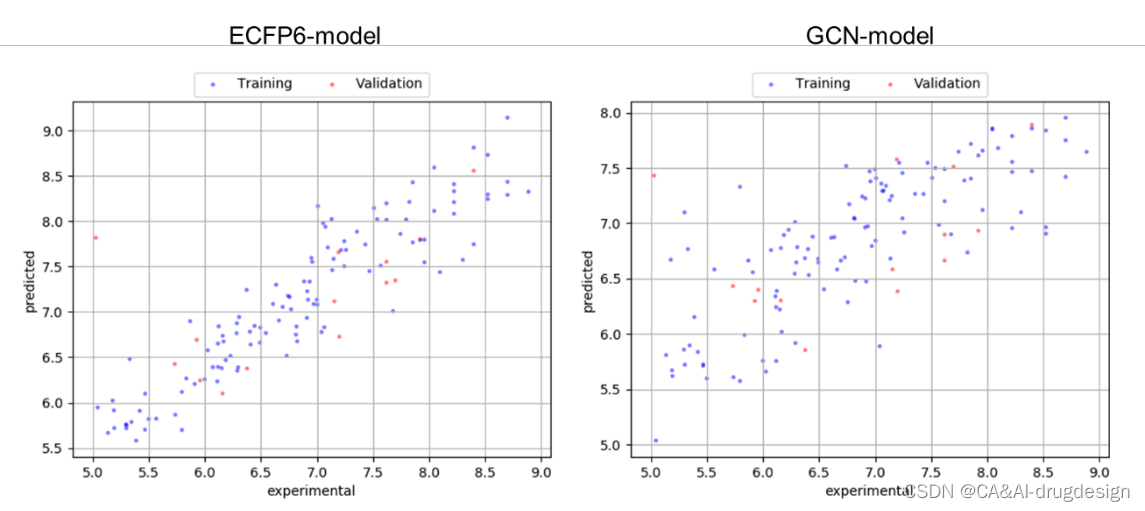
图S4 ECFP4模型(左)和GCN模型(右)的训练和验证集的预测与实验数据的散点图,来自肾素数据集。
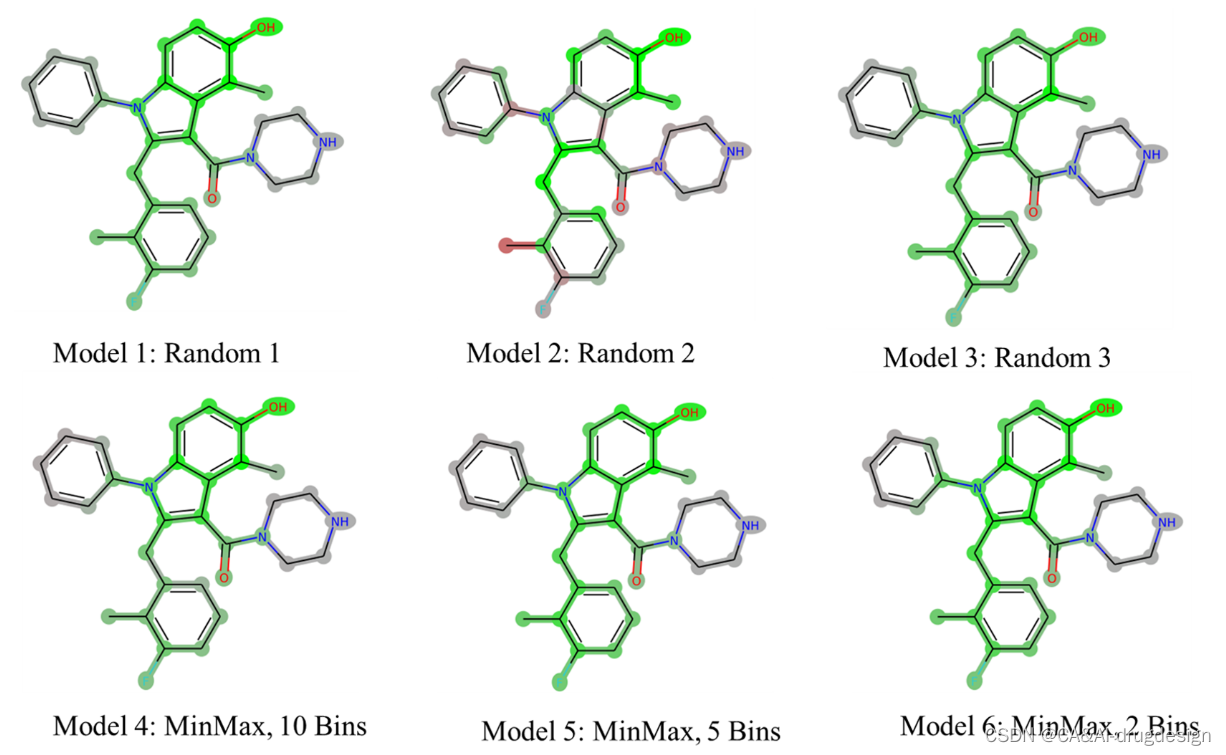
**为了了解不同数据集分割的影响,我们进一步研究了肾素数据集的预测性能行为及其对模型可解释性的影响。**我们使用MinMax算法划分数据集,结果表示,含有规模不断增加的测试集和验证集的新模型(见表3)并没有显示出预测性能的改善。接下来,通过不同的划分方法对训练、验证和测试集进行了六种不同的选择。对于每个选择,使用表1中列出的参考肾素模型的相同DNN参数,生成一个单独的基于指纹的模型。对于表3中的模型1-3,采用随机选择的验证和测试集。**对于模型4-6,首先将数据集按活性(pIC50值)分为10、5或2个规则的仓。**从每个活动仓中,使用MinMax算法(见上文)选择一个有代表性的子集,最后,在选择的验证和测试集中,通常会产生有代表性的活性分布覆盖。
表3. 基于指纹的模型在肾素数据集的训练集、验证集和测试集的不同分法中的表现
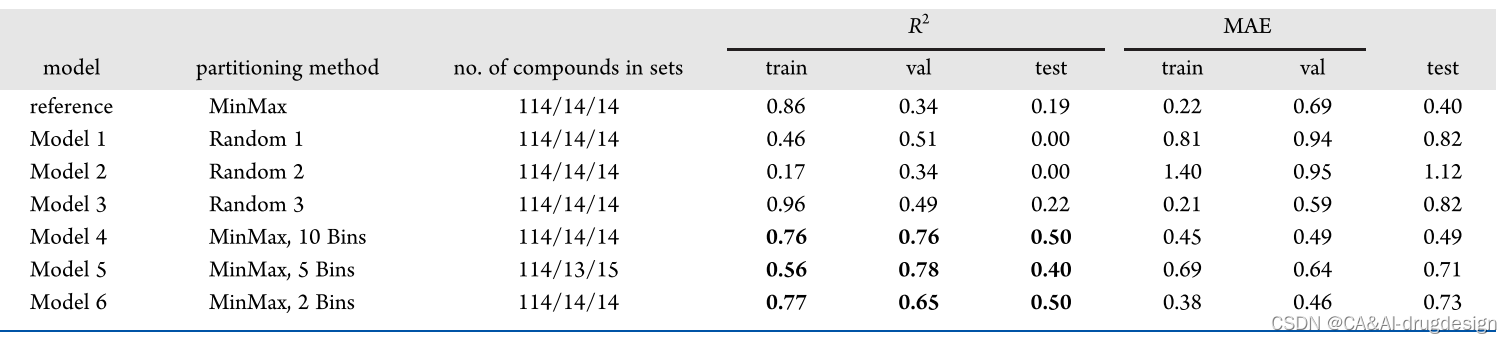
虽然从随机选择的验证集和测试集得出的模型1-3也未能为评价集产生可接受的R2值(测试集的R2值为0.00-0.22,见表3),但使用代表性覆盖活性分布的模型在测试集上的R2值在0.40和0.50之间,甚至对模型验证集显示出更好的性能。**这清楚地突出了不同评价集的选择对模型性能的影响。**尽管如此,这样的分区提供了一个非常乐观的模型评估方案,由于设计步骤对训练、测试和验证数据集进行了优化选择,因此具有最大的可实现的预测性能。因此,这项工作中大部分使用的原始模型对现实世界的情况提供了更真实的洞察力。**由于数据集规模和多样性有限,基于时间序列或骨架序列的验证策略无法应用。**作为最后一步,我们随后调查了这六个单独模型的最佳解释方法的性能(见下文)。
解释模型
在下一节中,我们比较了分别使用这两个靶标解释SAR趋势的模型预测的不同方法。我们表明,对于所有的数据集,结论是一致的。我们将强调不同方法解释的差异,并讨论为什么我们认为SHAP方法对我们的应用是最好的。**然而,"可解释性 "严格来说并不是一个可测量的数量。**因此,在这种情况下,对某一方法的有效性可能会有不同的意见。我们评估这些方法的目标是应用全面的和可理解的可视化。
解释肾素的多层(指纹)网络
对于使用指纹作为输入的网络模型,从技术角度来看,上述所有解释方法都可以应用。一般来说,所有基于SHAP的方法所提供的解释都被认为是非常相似的。尽管对于那些对亲和力显示出最大影响的指纹位来说,观察到了一些差异,但对于所产生的可视化热图中的累积atom wise的相关性来说,几乎没有差异(参见图5)。由于GradientSHAP和Deep- SHAP的运行速度明显快于KernelSHAP,在我们看来,这两种方法都是比较好的,特别是对于较大的数据集。因此,我们在下面的讨论中重点讨论DeepSHAP作为肾素的代表性解释方法的分析。
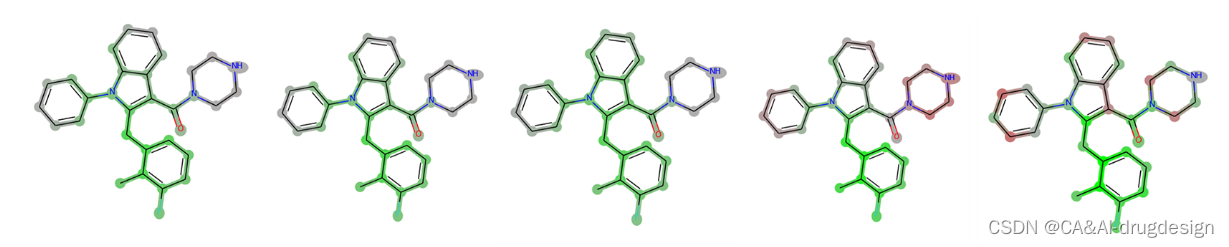
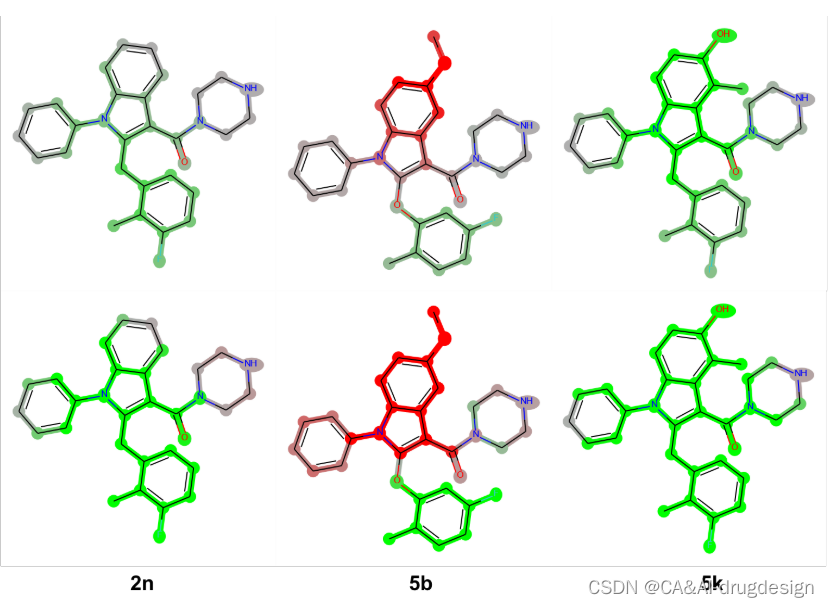
图5. 肾素吲哚-3-甲酰胺系列化合物5k的指纹模型的热图与截止点。从左到右,使用了以下方法:DeepSHAP、GradientSHAP、KernelSHAP、Gradient Heatmaps和Dummy Atoms。可以看出,所有三种SHAP方法产生的结果几乎是相同的。梯度热图的标记如描述的那样强,并受到分子活性的影响。在最后一张图片中,可以看到假原子方法的强烈局部解释的特点。
来自肾素数据集的三种吲哚-3-羧酰胺的代表性DeepSHAP热图,即化合物5k(PDB:3oot,IC50=0.002 μM,pIC50=8.70,pred. 9.15)、化合物2n(IC50=0.009 μM,pIC50=8. 05, pred. 8.12)和化合物5b(IC50=1.350 μM, pIC50=5.87, pred. 6.90),与实验检测的结合亲和力和基于指纹描述符的DNN模型预测的结合亲和力一起显示在图6。尽管图6中的这些化合物在结构上非常相似,但它们表现出明显不同的生物活性。
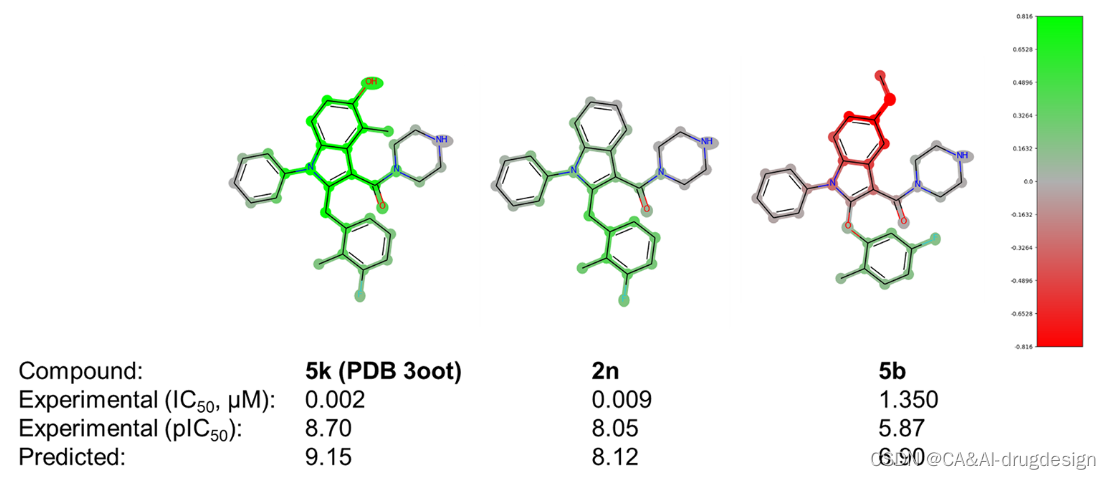
图6. 肾素吲哚-3-甲酰胺系列的三个分子的DeepSHAP解释。所有分子的实验的生物活性和预测的生物活性都在下面标明。片段亮点的颜色范围**从绿色(对提高结合亲和力有很好的贡献)超过灰色(没有明显的贡献)到红色(有很不利的贡献)。**这些解释有力地证明了与生物活性差异相对应的结构区域。
化合物5k(PDB:3oot,图6,左)是该图中活性最强的衍生物,这可归因于中心吲哚在C4(甲基)和C5(羟基)位置的合适修饰。**正如X射线结构3oot所概述的那样,5-OH基团参与了与His287形成有利H键,而4-甲基则与Thr77侧链甲基接触。**这两个基团在模型解释中相应地被标记为绿色,连同整个吲哚系统,表明在模型中对提高结合亲和力有非常有利的贡献。一般来说,较少的绿色表示在DNN模型中某一特定片段对结合亲和力的有利贡献较少。这种解释表明,在这个系列中,C4和C5周围的区域与亲和力高度相关。
这一解释在比较5k(左)和结构相似的类似物2n(中)时得到了强调。2n在吲哚的C4和C5位置缺乏取代物。这种修改使结合亲和力降低了4.5倍(IC50=0.009 μM),与图6中的模型解释很一致。现在,吲哚环只对整个结合亲和力的预测略有贡献,正如相应片段的绿灰色所显示的。相应的模型解释再次强调了在吲哚C5位置添加像甲氧基这样的不利取代基的负面作用,例如,对于化合物5b(IC50 1.350μM,图6,右)。通过对X射线结构的检查,**这个区域只允许有H键供体潜力的小的、极性的基团。**较大的基团很可能与该区域内笨重的His287侧链发生冲突,从而降低了亲和力。因此,C5-甲氧基基团的红色片段和连接的吲哚-苯并核心清楚地标志着这种取代模式对亲和力是不利的。
吲哚位置上的取代基C2通过最佳地填充疏水的肾素S1子口袋,也显示出对生物亲和力的明显影响。在先导化合物优化过程中,这个位置同时用苯氧基和苯甲基取代基进行装饰。图6中肾素数据集的DeepSHAP解释表明,苯甲基连接物(-CH2而不是连接到吲哚C2的-O)对亲和力的影响略微有利,这一点由绿色的苯甲基的-CH2基团显示。此外,从图6中的所有三个热图来看,指向Val30和Gly217附近的小子口袋的正甲基取代基对亲和力的有利影响是很明显的。在这里,整个数据集包含了更多的芳香族和脂肪族的变化,探索这个口袋的不同大小和静电特征。然后在Meta位置添加另一个氟取代基对亲和力再次显示出有利的影响,正如化合物2n所显示的那样。从图6的DeepSHAP热图(C2处绿色的芳香环)和肾素X射线晶体结构3oot中相应的有利的π-π相互作用(见上文)可以看出,整个苄基或苯氧基芳香环对结合亲和力有贡献。后者的相互作用在图4中用青色虚线表示。除了这种有利的作用外,5k(左边)和2n(中间)中的正取代基和Meta取代基也都用绿色突出显示,从而证实了它们在模型解释中的重要性。有趣的是,化合物5b中的正甲基和正氟取代基(图6,右)也被染成了绿色。这清楚地表明,对该分子5b的亲和力的关键不利因素是吲哚-5-甲氧基取代基。这一重要的结构-活性发现在参考文献41提供的SAR数据中得到了明确的强调。将预测和解释与该系列的其他分子进行比较时,结果变得更加清晰(见辅助资料图S5和S6)。这里显示了在吲哚-3-甲酰胺系列背景下的另外六个分子。在图S5中,模型正确地预测了下部苯环上的取代是不太有利的,另外还标记了吲哚C4和C5位置上缺少的取代基。同样,在图S6中,所有三个分子的吲哚-5-甲氧基取代基都被标记为红色,在分子5b中也可以看到。重要的o-甲基-m-F-苯取代基在所有三个分子中都被标为绿色,尽管其活性总体上相当低。这清楚地表明了这种可视化对药物设计的影响,因为重要的R-基团可以被识别。
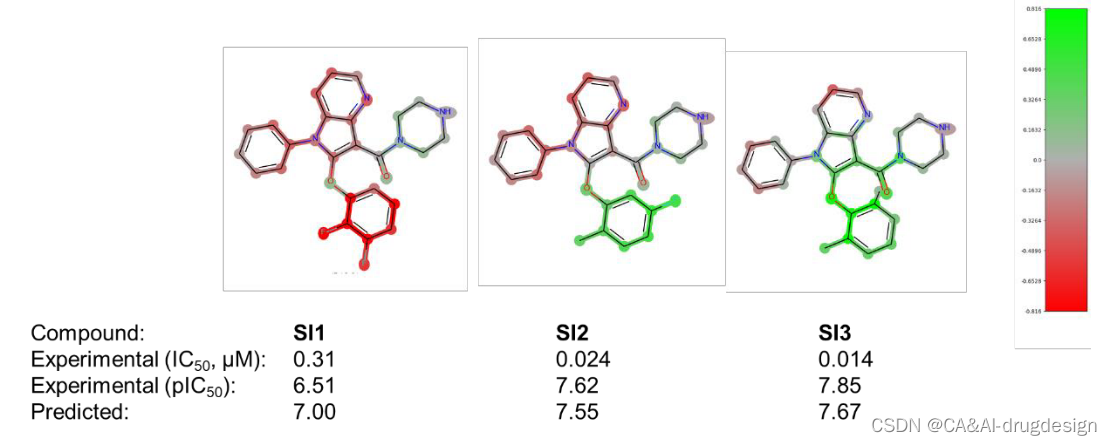
图S5 肾素吲哚-3-甲酰胺系列的三个分子的DeepSHAP解释(1)。所有分子的实验的生物活性和预测的生物活性都在下面注明。片段亮点的颜色范围从绿色(对提高结合亲和力有很好的贡献)超过灰色(没有明显的贡献)到红色(有很不利的贡献)。
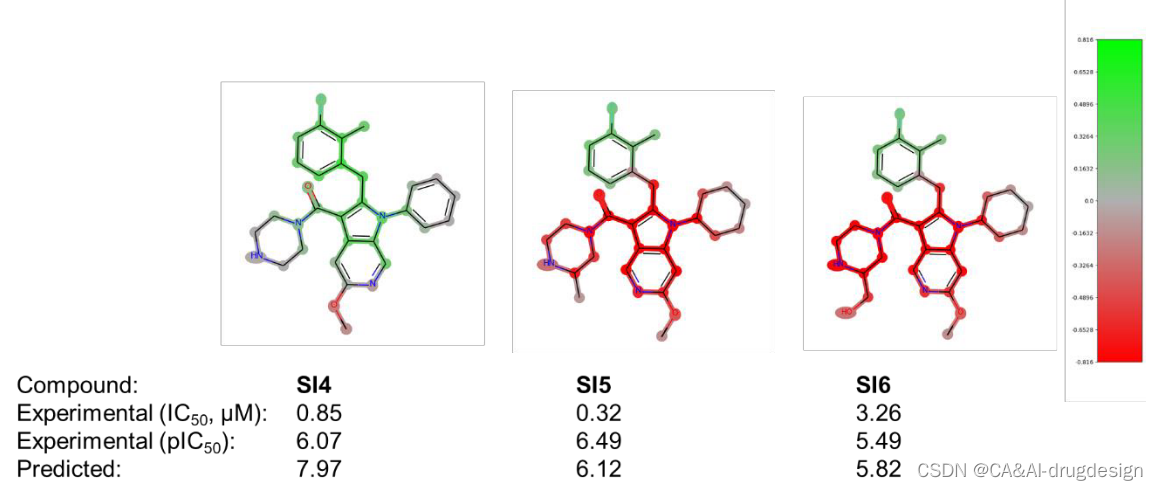
图S6 肾素吲哚-3-甲酰胺系列(1)的三个分子的DeepSHAP解释。所有分子的实验和预测的生物活性都在下面注明。片段亮点的颜色范围从绿色(对提高结合亲和力有很好的贡献)超过灰色(没有明显的贡献)到红色(有很不利的贡献)。
值得注意的是,在吲哚的C3位上的哌嗪基(参见图4)没有被这种解释方法突出显示出来(灰色),尽管它对肾素X射线结构的高结合亲和力非常重要。事实上,一个带正电荷的胺与天冬氨酸Asp32和Asp215的相互作用对大多数已知的肾素抑制剂系列是必不可少的。然而,如前所述,在数据集中没有包含明显的变化。对于所有的机器学习方法,由于缺乏结构和描述符的变化,导致无法识别与亲和力相关的子结构。因此,我们认为任何模型解释方法都不能突出这个分子的相关性。
因此,尽管与完整的SAR有冲突,数据集的依赖性是一致的,因为这种 "一般SAR知识 "也整合了来自其他来源的学习。这清楚地表明,来自机器学习模型的解释只与特定的模型有关,并与数据集及其结构和描述符的变化有关。
图6中显示的热图中另一个有趣的效果是具有不同生物活性的三种不同分子的吲哚支架本身和连接原子的颜色差异。这种差异可以通过考虑DNN模型的输入而合理化。在这个案例中,使用ECFP6型指纹来建立DNN模型,任何与亲和力有关的子结构模式都可以覆盖输入分子的很大一部分,如果该原子位于子结构的另一端,那么它与可能直接影响活性的原子的距离可达6个键。因此,受一个取代物影响的拓扑结构通常是整个结构的较大部分,而不仅仅是其近邻的部分。
如上所述,我们随后按照验证集和测试集选择的不同方法研究了替代模型,以及它们对肾素数据集的模型可解释性的影响。统计结果总结在表3中,而使用DeepSHAP对化合物5k的视觉解释显示在图7中。该图表明,尽管验证集和测试集的统计数据不同,但几乎所有来自随机选择(模型1-3)或设计的数据集(模型4-6)的模型都产生了类似的解释。活性的关键特征总是被DeepSHAP所识别。一个例外是来自随机选择的验证集和测试集的模型2(图7,中上部面板)的解释。在这里,连接到吲哚C2位置上的苄基的正甲基的影响没有被正确地捕捉到。此外,连接到吲哚N1的苯基的贡献往往过于消极。然而,这个特殊的模型在所有组别中显示出极低的R2值(表3),指出这是一个不可靠的模型。这也表明,具有这种性能的模型不应考虑进行解释。总之,所有具有可接受的或良好的统计数据的模型都产生了一致的解释,这清楚地表明,这种方法对于不同模型的建立条件是稳定的。
解释Factor Xa的多层(指纹)网络
对于fXa,我们采用了两个不同的数据集来建立和解释DNN模型。这样就可以比较,这些不同规模和化学多样性的数据集显示出来的狭窄或宽广的SAR,是如何影响DNN模型的解释性。表1和表2中总结了这两个模型的结果。
图8显示了吲哚-2-甲酰胺骨架上的一些信息性的fXa抑制剂。所有三个分子3、1和21从左到右显示出对fXa的明显不同的生物活性,这反映在模型预测中。一些重要的模型解释方面也可以从该图中提取。
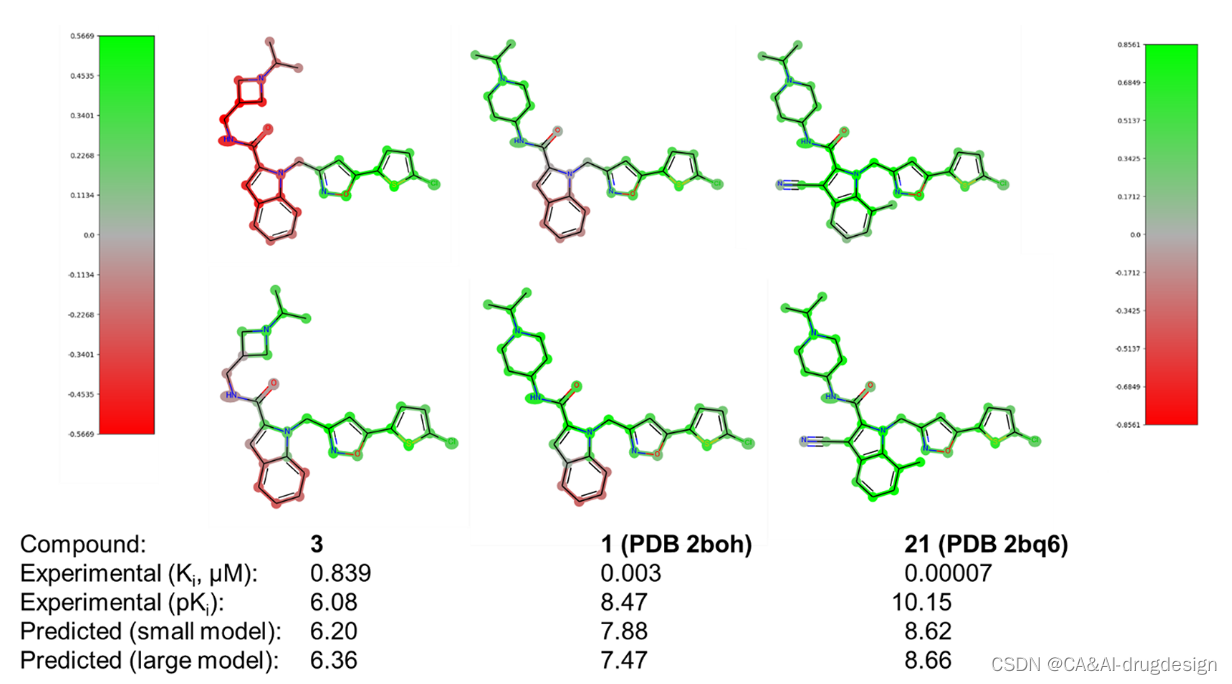
图8. 针对小型fXa数据集(顶行分子,左色条)和大型fXa数据集(底部分子,右色条)得出的DNN模型的DeepSHAP指纹热图。所有分子的实验和预测的生物活性都在下面注明。这些解释有力地证明了与生物差异相对应的结构区域。
与化合物1相比(PDB 2boh,图3和 图8中间部分),左边的fXa抑制剂(化合物3,实验亲和力:Ki=0.839 μM,pKi 6.08,预测值6.20(小模型),6.36(大模型))被一个氮杂环取代,而不是指向蛋白酶S4口袋的较大的哌啶环。虽然两个结构的basic氮处的异丙基修饰是相同的,但我们认为化合物3在这个口袋中的匹配度不是很理想,蛋白质与配体之间的相互作用也不是很有利。此外,所有分子中带正电的氮都参与了几何学上敏感的阳离子-π相互作用。在含杂氮环丁烷的化合物3中,这种阳离子-π的相互作用显示出略微不那么有利的几何形状,这可能也是造成活性差异的原因。这两种效果都清楚地反映在图8(左)的视觉解释图中,特别是来自小型fXa数据集的DNN模型。
虽然两个DNN模型在亲和力预测方面表现相似,但它们产生的解释略有不同(图8,中间部分)。特别是,从较大的fXa数据集中得到的DNN模型的热图中,负贡献(红色)不太明显。这是因为与涵盖单一优化系列的较小的fXa数据集相比,较大数据集的模型是用各种不同的分子建立的,其中高活性抑制剂的比例较低。**由于所有的SHAP解释总是参考所采用的数据集的平均值,这个 "baseline "对来自较大数据集的模型来说是较低的。**这就导致在小数据集的DNN模型中具有负相关性的一些片段,在大数据集的模型中表现出更多的中性贡献。因此,图8(左)中的化合物3不再被认为是相对于这个转移的baseline来说特别 “无活性”。此外,在大数据集的热图中,负的热图贡献更多集中在这个分子中特别出现的原子上,而三级氮仍然被认为是一个重要的活性特征。
fXa抑制剂化合物1(PDB:2boh,图8中部 与化合物3相比,其活性更高(实验亲和力:Ki=0.003 μM,pKi 8.47,预测值7.88(小模型),7.47(大模型)),这可以归因于哌啶侧链对蛋白酶S4口袋显示出良好的立体和静电互补性(参照图4)。在检查两个DNN模型中该化合物的热图时,模型预测反映了这一点。然而,这些热图表明,中间未修饰的吲哚支架还不是实现非常高的fXa亲和力的最佳选择。同样,对于这个分子,DeepSHAP得出的热图的不同baseline的影响变得明显。特别是,较小的fXa数据集包含许多具有额外有利的吲哚取代基的分子,这就产生了一些未被取代的支架的负贡献。这一点在较大的fXa数据集中也不太明显。
最后,fXa抑制剂化合物21(PDB:2bq6,图8右面板)显示出明显较高的fXa活性(实验性亲和力:Ki=0.00007 μM,pKi=10.15,预测值8.62(小模型),8.66(大模型))。这种非常高的结合亲和力是由于中心吲哚的两个有利的取代,分别为第3位的氰基和第7位的甲基。在基于结构的设计指导下,这种特殊的抑制剂是由这两个单独的取代物组合而成的。如图9所示,疏水的7-甲基填补了吲哚C7原子和Gln192侧链的脂肪族部分之间的空间。3-氰基取代基位于glu217侧链的顶部,取代了fXa/1复合物中的一个结构保守的水。这个氰基取代基参与了与glu217的脂肪族碳的有利的相互作用和更有利的静电相互作用,如邻近的Arg222侧链的胍。这些有利的取代基在图9(右)的两个视觉解释图中都被清楚地突出显示为有利的,对两个DNN模型而言。
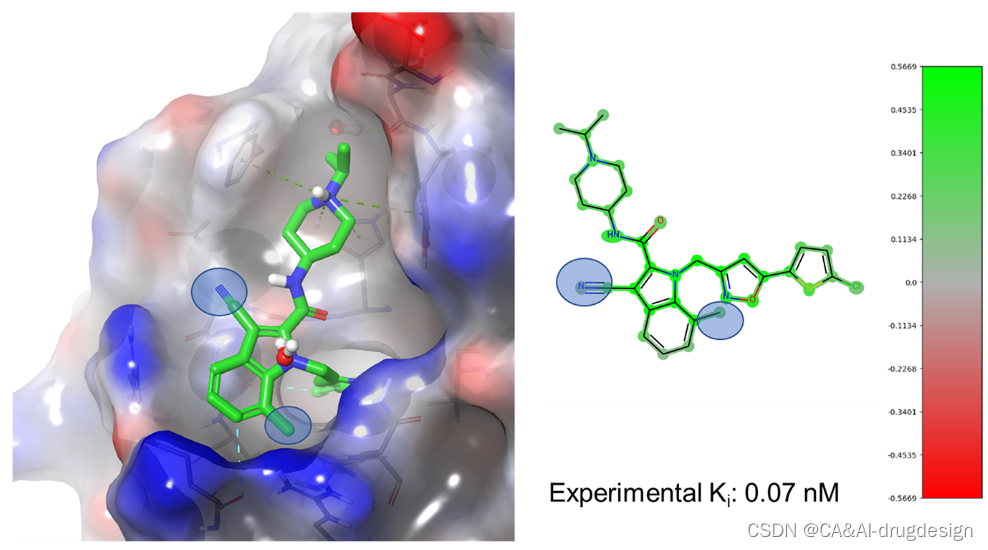
图9. 左图:吲哚-2-甲酰胺21(绿色碳原子,3.0 Å分辨率,PDB:2bq6,Ki=0.07 nM)与人类factor Xa复合物的X射线晶体结构。灰色的碳原子显示了蛋白质的结合部位。此外,蛋白质结合点被显示为一个可接触溶剂的表面,其上映射着静电电位(蓝色=正,红色=负表面区域)。更多细节见图3。右图:化合物3的DeepSHAP指纹热图和针对小型fXa数据集的DNN模型的颜色条。
这些例子表明如果可以开发一个预测性的局部DNN模型,DeepSHAP与基于ECFP指纹的DNN模型相结合,有可能解释化学系列的SAR,并为进一步优化提供指导。**另一方面,结合一个靶标的所有配体信息的全局模型加解释将对探索命中化合物和重新设计骨架更有用,因为这些模型捕捉到非常普遍的SAR趋势和决定亲和力的结构条件。**正如我们所表明的,监测所采用的数据集中的活性分布是非常重要的,因为这决定了这种方法的参考baseline,所有的片段解释都要考虑到这一点。
另一个类似的影响与模型的预测性能有关。由于解释是相对于数据背景进行的,因此正确预测化合物之间的个别相对趋势是很重要的。如果两个非常相似的分子的亲和力差异被DNN模型预测为不正确的趋势,那么模型的解释也可能反映这种不正确的趋势。图10显示了fXa X射线结构2boh(化合物1)的另外两个类似物,即化合物4(左边,实验活性:Ki=0.003 μM,pKi 8.50,预测:7.25)和化合物6(右边:实验活性:Ki=0. 023 μM,pKi 7.64,预测值:7.60)。由于位于蛋白酶S1口袋的苯环上有一个碘原子,所以化合物6在实验中的活性较低,而活性较高的分子在这里含有一个溴。早期的研究表明,卤素原子的类型和大小对Tyr228的有利的卤素-π作用有明显的影响。尽管碘的特点是与π电子系统相互作用的典型σ孔,但它太大了,无法最佳地融入这个口袋,这使活性降低了7倍∼。
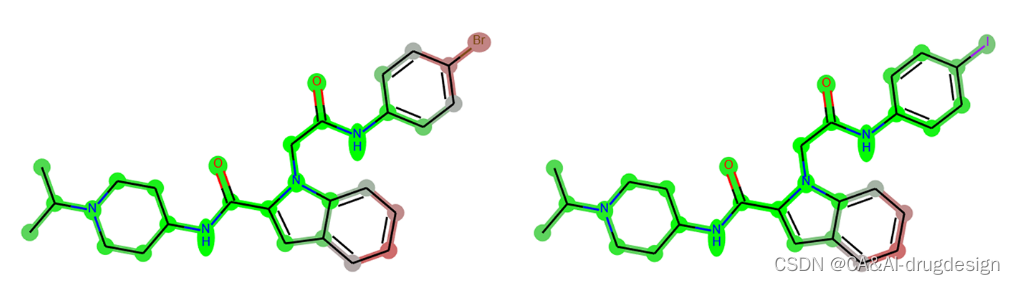
图10. 根据大数据集建立的DNN模型对两种fXa抑制剂的解释。左图:化合物4,实验活性:Ki=0.003 μM,pKi=8.50,预测值:7.25;右边:化合物6,实验活性:Ki=0.023 μM,pKi=7.64,预测值:7.60。
然而,当考虑到来自DNN模型的预测时,实验中的活性趋势没有被重现。相反,含碘结构被错误地预测为更有活性。在这里,模型的解释与预测一致,并突出了碘原子对亲和力更有利的贡献(即绿色)。另一个例子是Supporting information图S6中的分子SI4。活性显然被预测得太高了。然而,该分子的活性仍然不如图5中的主导化合物5k。即使在这种情况下,甲氧基的正确影响仍然被正确解释。
其他解释方法的表现
使用梯度热图法得到的DNN模型解释与各种SHAP方法的解释非常相似。一个重要的观察结果是,从梯度热图中获得的解释的数值与预测的大小相关联。因此,例如在某些情况下,对一个无活性分子的解释会产生较高的数值,导致较高的差异而没有明显的原因(参见图11)。
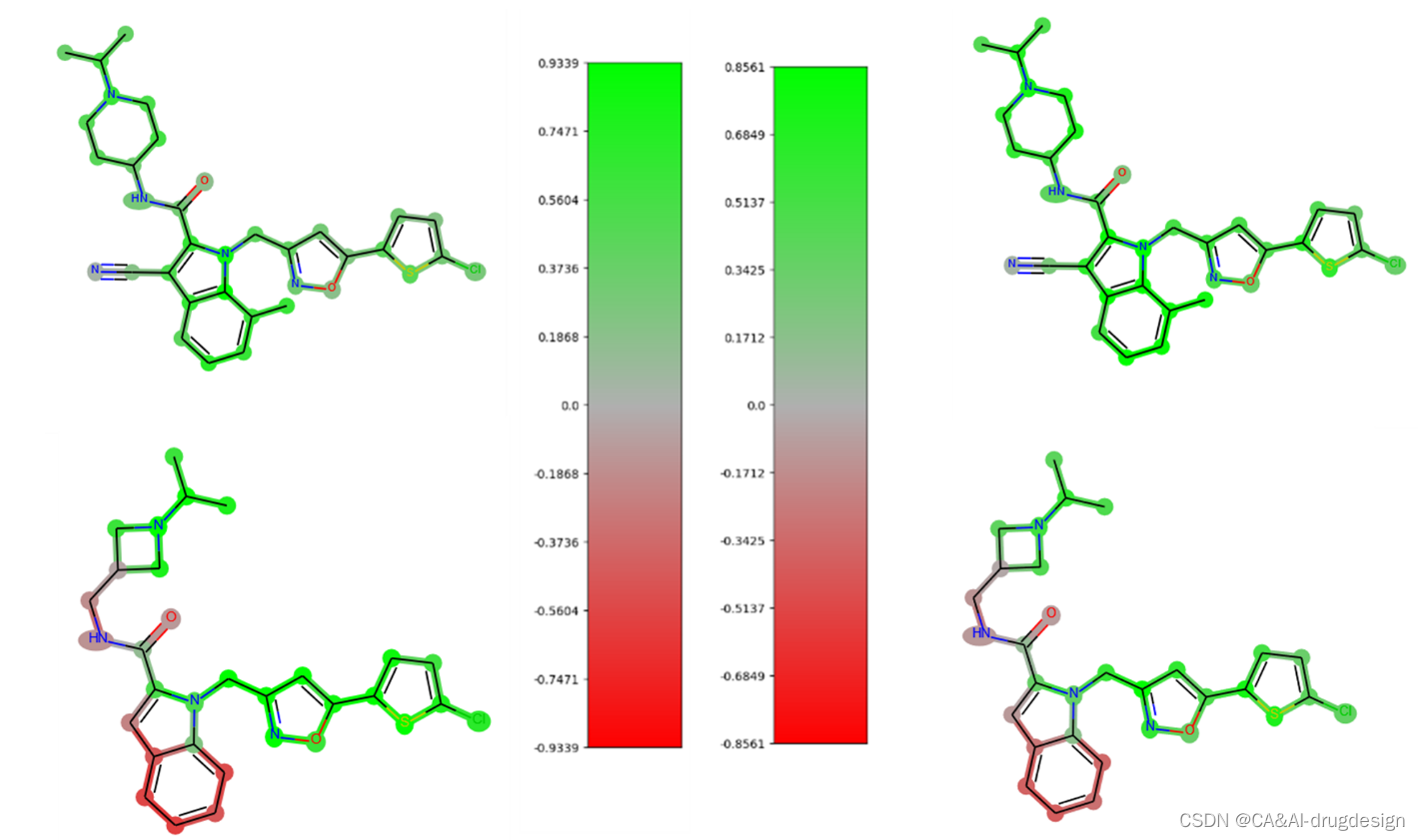
图11. 左图:Factor Xa抑制剂21(上)和3(下)的梯度热图。右图:抑制剂21(上)和3(下)的DeepSHAP解释。所有的热图都是使用DNN模型从大型fXa数据集中得出的。
图11显示了两个Factor Xa抑制剂的梯度热图(左)和DeepSHAP解释(右),即化合物21(上图)和3(下图)。这些热图是使用DNN模型从大型fXa数据集中得出的。梯度热图的颜色比例一般产生较大的数值。此外,在梯度热图中,活性较低的化合物3的亮点明显更强。虽然这个特定的热图本身是一致的,但它与这个化学系列的其他热图缺乏合理的可比性,因为较高的数值并不一定与分子中变化的重要性相关。这意味着SAR趋势不能始终如一地从一个较小的系列中提取相关分子的相应热图,这使得新分子的指导性设计更加困难。
虽然使用Dummy Atom方法也可以得到相关的解释,但详细的结果总是不那么令人满意。**特别是,解释更多的是围绕特定原子的局部,但不一定以可靠的方式从已知的SAR中突出相关区域。**此外,有时发现无法区分的原子会产生明显不同的解释,这无助于提高该方法的可信度。这些无法区分的原子指的是由于对称性,在结构拓扑中无法被区分的原子。例如,由于旋转对称性,这可能是苯中的所有六个原子。有趣的是,在最近的一项研究中,在一大组实验确定的活性悬崖上研究了使用假原子进行特征归属的情况。在识别归属特征的正确符号方面,结果优于其他特征归属技术。然而,分析中忽略了特征归属的大小。
通过LRP(分层相关性传播)获得的解释也不符合其他方法观察到的解释质量。这种方法适用于特定神经网络的输出,并通过网络将其传播回去,从概念上看,这似乎是一种有趣的方法。因为我们观察到数值不稳定,进一步优化实现可能有助于获得更好的结果。图12给出了从LRP解释中获得的结果的一个例子,可以看出,解释的最高贡献与特定的特征无关,而是散布在分子的各个部分。其他模型和分子也有类似的情况。因此,我们决定,LRP方法并不完全适合我们目前的目标。更详细的分析可能会揭示更多的细节。然而,由于我们已经观察到基于SHAP的方法表现非常好,我们没有进一步研究这种方法。
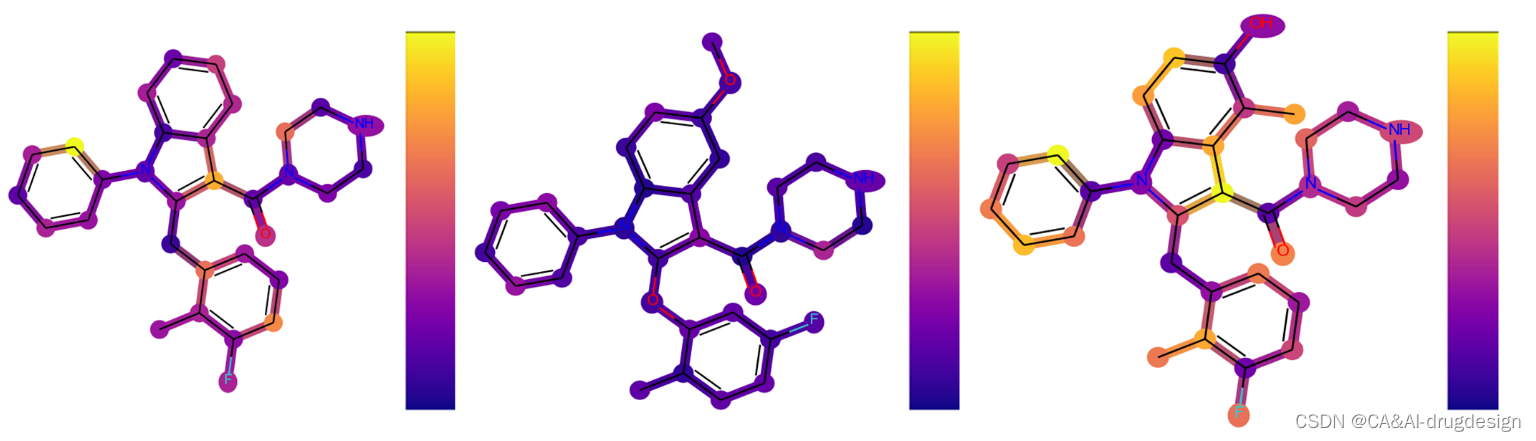
图12. 用LRP为GCNN模型生成的热图。最高的贡献并不总是与最相关的特征相联系,而且在同一系列的多个分子中并不稳定,因此严重限制了设计项目的适用性。只有正面的贡献可以被提取出来,范围从0(蓝色)到黄色。
解释图卷积神经网络
对于GCNN来说,我们对模型解释的结果总的来说是不太乐观的。GCNN解释的一般问题是,它们不使用典型的统一输入格式,这使得标准方法的应用变得困难,需要定制实现。特别是,一个原子的每个特征都会产生一个解释,这使得可视化更具挑战性。此外,其复杂的结构使方法的应用更加困难,因为各层往往包含大量的操作,需要加以考虑。从图13可以看出,正负贡献或多或少地分布在所有分子上。因此,不能提取有关化学特征的信息。
图13. 由GCNN模型生成的梯度热图。从左到右:贡献的正值之和;贡献的负值之和;正值和负值之和映射在一起;绝对值的总和。可以看出,基本上不能提取任何信息,因为所有的原子都有负的和正的特征,而且差异与分子结构上的重要区域没有关联。
虽然baseline Dummy Atom方法产生了最好的解释,即使这些解释在大多数情况下与我们靶标的已知SAR不一致。因此,它们对指导后续化合物设计周期的作用是有限的。对这一领域的进一步分析和研究(特别是将GCNN的解释映射到基于原子的热图上)是未来研究的一个非常有趣的课题。
其他可以应用于基于GCNN模型的方法是梯度热图和LRP。**这两种方法都已经成功地应用于GCNN,但我们无法重现这些有希望的结果(见图13的例子)。**这方面的一个主要因素是与我们研究中采用的不同的GCNN架构有关。DeepChem GCNN架构更加复杂,特别是包含大量的 "最大 "操作。这可能会导致梯度热图方法的梯度计算问题,并可能解释不令人满意的结果。虽然LRP不存在这个问题,但如前所述,我们的结果可能会通过不同的技术实现而得到改善。
结论
我们的研究清楚地表明,使用指纹向量作为描述符的基于神经网络的模型可以被有效地解释。从测试的方法来看,所有单独的SHAP变体和梯度热力图在较小的程度上被认为适合于捕捉所评估的数据集中配体系列的基本SAR趋势,这与一般的SAR知识和X射线晶体结构信息相对应。这些解释可以通过热图收集并全面呈现,这将立即激发具有前瞻性的药物化学家和计算化学家的创造力,**思考特定基团的潜在替代物或电子等排体。**这些解释清楚地突出了分子的哪些部分被认为对解释亲和力或任何其他目标特性是有利的或不利的。与基于指纹的方法相比,GCNN方法的优点是在模型优化过程中学习了分子表征。**由于也能够同时解释GCNN模型,这将使得该模型能呈现更详细的解释。此外,这些模型通常比基于指纹的模型显示出更好的性能,因此这些模型的解释性将允许将更好的模型整合到工作流程中。**然而,我们对基于GCNN预测的解释性和说明性的探索不太令人满意,可能需要进一步研究如何解释GCNN模型以指导药物设计。未来的研究可以包括基于SHAP的对模型进行全局解释,这可以使人们对整个数据集和模型有更好的理解。
所有的解释一般都需要关于靶标、数据集和可能的三维蛋白质-配体结构的 "背景 "信息,因此不应该作为单独的 "图片 "来分享。特别是,用于建立模型的数据集的背景对于理解解释的适用性至关重要。因此,一个对数据和DNN模型有足够了解的科学家应该为了可解释性对信息进行预处理,以提供一个合适的背景。模型错误是解释的一部分,这一事实也应该被考虑。
**我们认为这些热力图是一个有价值的工具,可以促进与其他项目组成员交流统计结果。**特别是,解释模型决定的能力可能会增加对机器学习技术的普遍接受度,如果谨慎解释的话,甚至有可能被用来获得新的机制洞察力。这是一个非常有价值的前景,因为只有在一个环境中,从机器学习中获得的数据被完全整合到药物设计周期中,才能充分地利用模型的能力,并能指导设计思路,**例如,将来自不同分子的两个正面解释的R-基团组合成一个新的化合物。**此外,可解释的模型允许以无偏见的方式评估预测质量和改进模型。
未来研究的另一个课题是不同的数据集拆分如何影响模型创建的解释。特别是,使用更复杂的验证策略,如聚类拆分验证和时间序列拆分验证的效果,将是值得观察的。最终,这些方法必须应用于一个前瞻性的场景,在这个场景中,解释的价值可以得到真正的检验,但这是目前这项工作的范围之外。
版权声明:本文内容由互联网用户自发贡献,该文观点与技术仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容, 请发送邮件至 dio@foxmail.com 举报,一经查实,本站将立刻删除。

