

相关性:一对个体的遗传相关性强度。传统的GWAS假设所有的受试者都是不相关的,即没有一对个体比二级亲属关系更密切(三代内)。如果没有适当的校正,包含亲属可能会导致对SNP效应大小的标准误差的估计有偏差。
#检查你分析的数据集是否有神秘的相关性是很重要的。
#假设是一个随机总体样本,在本教程中,我们将排除所有超过pihat阈值0.2的个体。
#查看pihat>0.2的个体对。--genome调用IBS/IBD计算,并且之后在plink.genome文件中写入报告。--min删除PI_HAT值低于给定截止值的行。
plink --bfile HapMap_3_r3_10 --extract indepSNP.prune.in --genome --min 0.2 --out pihat_min0.2

less pihat_min0.2.genome
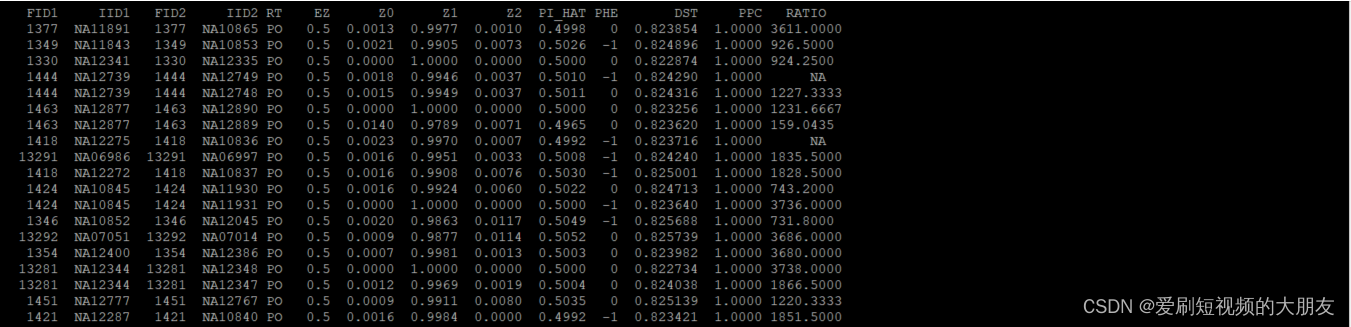

第一列为个体对中第一个个体的FID,第二列为个体对中第一个个体的IID,第三列为个体中第二个个体的FID,第四列为个体对中第二个个体的IID,第五列为从.fam或.ped文件中推断出的相关类型,第六列为IBD共享预期值,基于.fam/.ped的关系,第七列到第九列为P值,第十列为IBD占比(例如:P(IBD=2) + 0.5P(IBD=1)),第十一列为成对表型代码(1,0,-1 = case-case, case-ctrl, ctrl-ctrl对),第十二列为IBS距离,即(IBS2 + 0.5IBS1) / (IBS0 + IBS1 + IBS2),第十三列为IBS 二项分布检验,第十四列为HETHET: IBS0 SNP比值(期望值2)


#现在我们将再次寻找具有pihat >0.2的个体。
plink --bfile HapMap_3_r3_11 --extract indepSNP.prune.in --genome --min 0.2 --out pihat_min0.2_in_founders
#“pihat_min0.2_in_founders.genome”显示,在排除所有非创始人后,HapMap数据中只剩下1对pihat大于0.2的个体。

#对于每一对“相关”个体,pihat >0.2,我们建议取消呼叫率最低的个人。
plink --bfile HapMap_3_r3_11 --missing
#在我们的数据集中,个体13291 NA07045的呼叫率较低。
vi 0.2_low_call_rate_pihat.txt
i
13291 NA07045
# Press esc on keyboard!
:x
# Press enter on keyboard
#删除具有pihat>0.2的“相关”对中呼叫率最低的个体
plink --bfile HapMap_3_r3_11 --remove 0.2_low_call_rate_pihat.txt --make-bed --out HapMap_3_r3_12
#恭喜你! !您刚刚成功地完成了第一个教程!你现在可以进行一个适当的基因QC。
#在下一个教程中,使用脚本:2_Main_script_mds.txt,你需要以下文件:
- The bfile HapMap_3_r3_12 (i.e., HapMap_3_r3_12.fam,HapMap_3_r3_12.bed, and HapMap_3_r3_12.bim
- indepSNP.prune.in
版权声明:本文内容由互联网用户自发贡献,该文观点与技术仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容, 请发送邮件至 dio@foxmail.com 举报,一经查实,本站将立刻删除。

